1-8 of 8 results
-

Expert Panel at ISAPP Annual Meeting Addresses Probiotic Use for Premature Infants
The use of probiotics in premature infants has been highly topical in recent months. Probiotic use for the prevention of… -
Probiotic Administration in Preterm Infants: Scientific Statement
Board of Directors, International Scientific Association for Probiotics and Prebiotics in collaboration with Dr. Geoffrey Preidis MD PhD, Pediatric Gastroenterology,… -

EFSA’s QPS committee issues latest updates
By Bruno Pot, PhD, Vrije Universiteit Brussel and Mary Ellen Sanders, PhD, Executive Science Officer, ISAPP On July 2nd, the… -
The FDA’s view on the term probiotics, part 2: Further down the rabbit hole
By James Heimbach, Ph.D., F.A.C.N., JHEIMBACH LLC, Port Royal, VA A number of weeks ago I wrote on the ISAPP… -

The FDA’s view on the term probiotics, part 1
Over the past 20 years as a food and nutrition regulatory consultant, I have filed about 40 GRAS notices with… -

Challenges ahead in the probiotic field – insights from Probiota 2019
By Dr. Mariya Petrova, Microbiome insights and Probiotics Consultancy (MiP Consultancy), Bulgaria. Recently, I attended the Probiota Conference, which brings… -

FDA/NIH Public Workshop on Science and Regulation of Live Microbiome-based Products: No Headway on Regulatory Issues
September 20, 2018 By Mary Ellen Sanders, PhD, Executive Science Officer, ISAPP On September 16, 2018, the US Food and… -
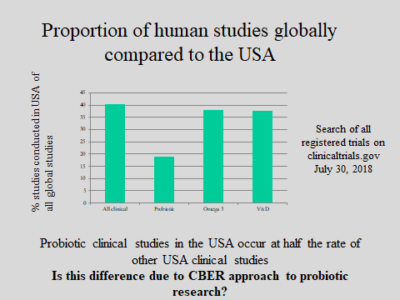
CBER to hold public workshop on regulation of biologics
FDA’s Center for Biologics Evaluation and Research (CBER) is convening a public workshop Sept 17 in Rockville MD on the…